MetaVelvet : An extension of Velvet assembler to de novo metagenome assembly from short sequence reads
Introduction
Motivation: An important step of "metagenomics" analysis is the assembly of multiple genomes from mixed sequence reads of multiple species in a microbial community. Most conventional pipelines employ a single-genome assembler with carefully optimized parameters and post-process the resulting scaffolds to correct assembly errors. Limitations of the use of a single-genome assembler for de novo metagenome assembly are that highly conserved sequences shared between different species often causes chimera contigs, and sequences of highly abundant species are likely mis-identified as repeats in a single genome.
Methods: We modified and extended a single-genome and de Bruijn-graph based assembler, Velvet, for de novo metagenome assembly. Our fundamental ideas are first decomposing de Bruijn graph constructed from mixed short reads into individual sub-graphs and second building scaffolds based on every decomposed de Bruijn sub-graph as isolate species genome.
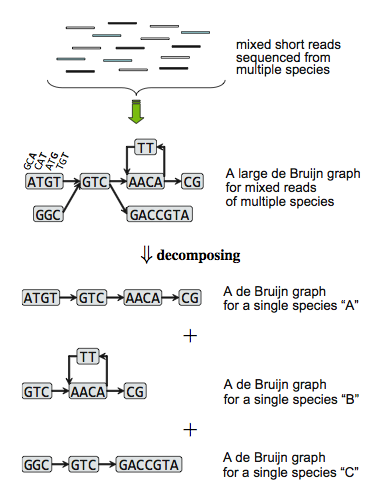
What is improved?
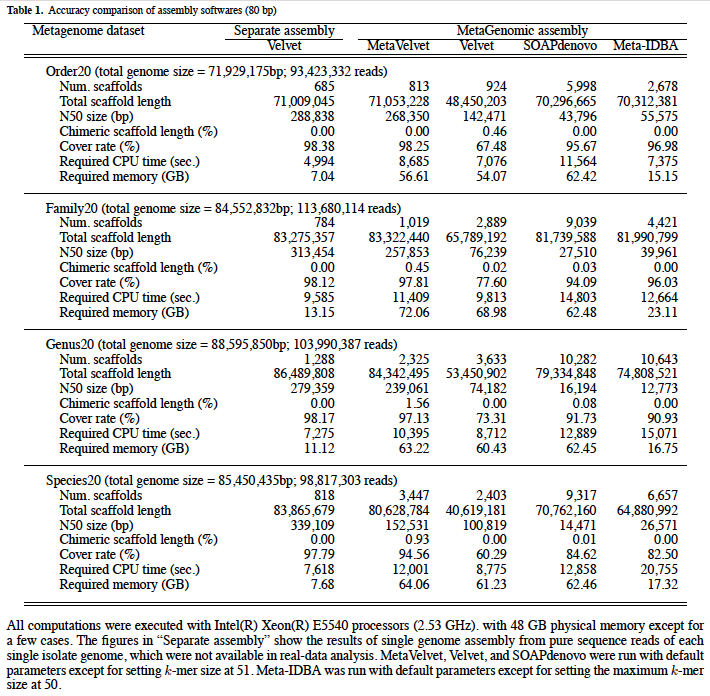
Longer N50 size: On artificially generated simulation datasets of short sequence reads, it is proven that MetaVelvet assembled metegenomic read data with significantly longer N50 sizes than single-genome assemblers (Velvet and SOAPdenovo) and another meta-genome assembler (MetaIDBA) as shown in the table below.
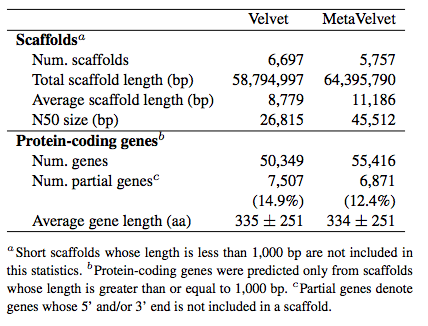
Improved gene prediction: We applied Velvet and MetaVelvet to real metagenomic dataset (SRR041654 and SRR041655; human gut microbiome data downloaded from NCBI sequence read archive). Then, MetaGeneAnnotator was used to predict a gene set of the microbiome. From the results, it is demonstrated that MetaVelvet can efficiently avoid chimeric scaffolds, resulting in longer scaffolds, increased number of predicted genes, and decreased fraction of partial genes.
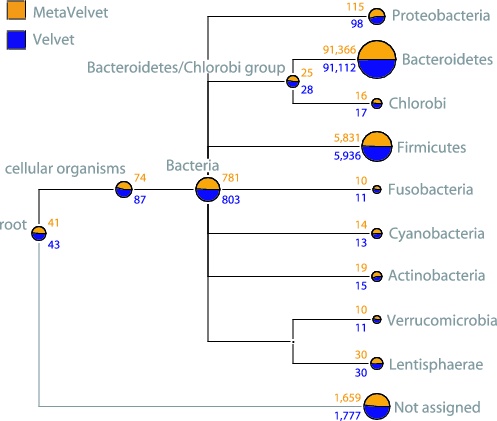
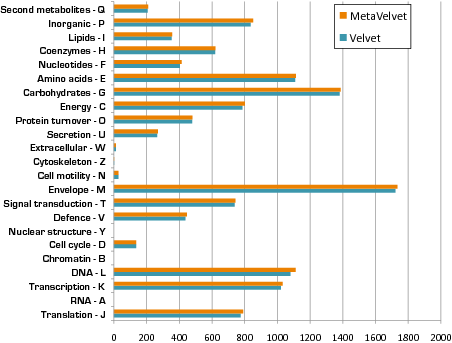
Widely applicable to matagenomic analyses: We also compared the results of metagenomic analyses, such as taxonomic content analysis, functional composition analysis, and pathway mapping analysis, between Velvet and MetaVelvet. The results demonstrate that taxonomic content can be estimated more clearly when based on the MetaVelvet scaffolds than when based on the Velvet scaffolds as shown in the figure below left (the number of genes that cannot be assigned to phylum-level taxonomy is substantially decreased). Moreover, the result of functional composition analysis also shows that MetaVelvet is more appropriate for metagenomics purpose than Velvet as shown in the figure below right (the number of genes assigned to any functional category is improved).
Source Code (Release Notes)
back to IndexThe following source codes is freely available under GNU General Public License.
- MetaVelvet-v1.2.01 (May.29,2012)
- enable to use paired-end information in a graph decomposition step.
- three options are newly added in order to specify how to use paired-end connections for graph decomposition: -valid_connections, -noise_connections, and -use_connections.
- for details, please see Advanced topic 3: how to use paired-end information for graph decompositoin. - MetaVelvet-v1.1.01 (Oct.19,2011)
- this version is based upon Velvet 1.1.06.
- fix a frequent bug: no peak detection error.
- fix a frequent bug: infinite loop error.
- modify the graph split algorithm: an exception for "knock-turn" loops.
- modify the graph split algorithm: split priority is now based on peak IDs rather than node IDs.
- MetaVelvet-v0.3 (Jan.14,2011)
- this version is based upon Velvet 0.7.62.
Source codes at these versions can be downloaded from here.
Requirements (>= 1.1.01)
- Required libraries:
- zlib library (>= 1.2.3) - zlib library is also included in the Velvet distribution.
- Required programs:
- velveth (>= 1.1.06) - included in the Velvet distribution.
- velvetg (>= 1.1.06) - included in the Velvet distribution.
- g++ (>= 4.3.2) - for compile.
- Recommened environment:
- OS : standard 64bit Linux (It can in theory function on a 32bit environment, but not recommended).
- RAM: at least 12GB (more is no luxury).
Install MetaVelvet
- Download source code from here, and decompress the tar ball.
~$ tar zxvf MataVelvet-v1.1.01.tgz
Alternatively, up-to-date source code is available from github MetaVelvet project.~$ git clone git://github.com/hacchy/MetaVelvet.git
- Change directory to the MetaVelvet directory.
~$ cd MetaVelvet-v1.1.01
- Compile MetaVelvet execution files.
~/MetaVelvet-v1.1.01$ make ['MAXKMERLENGTH = k'] ['CATEGORIES = cat']
k - the maximum k-mer size you like to use.
cat - the maximum number of read categories (i.e., maximum number of libraries in different insert lengths)
Then, an executable files, meta-velvetg will be created (>= 1.1.01). - Copy the two executable files to /usr/bin or a directory you like to install.
~/MetaVelvet-v1.1.01$ cp meta-velvetg /usr/bin/
Getting started (>= 1.1.01)
MetaVelvet (>= 1.1.01) uses velveth and velvetg outputs for metagenomics assembly. Three files ("Sequences", "Roadmaps", and "Graph2") are needed for running meta-velvetg.
- Download a small dataset from here, and decompress the tar ball.
~$ tar zxvf HMP.small.tar.gz
- Execute velveth to import read sequences and construct k-mer hash table.
~$ velveth out-dir 51 \
Please check that "out-dir/Sequences" and "out-dir/Roadmaps" files are created. Note that k-mer size has important effect on assembly results. If your short read data is longer than 65bp, we recommend k-mer longer than 51.
-fasta -shortPaired \
HMP.small/SRR041654_shuffled.fasta \
HMP.small/SRR041655_shuffled.fasta
- Execute velvetg to construct an initial de Bruijin graph.
~$ velvetg out-dir -exp_cov auto -ins_length 260
Please check that "out-dir/Graph2" file is created. Please use -exp_cov auto option, otherwise meta-velvetg cannot be executed. - Execute meta-velvetg to output scaffold sequences.
~$ meta-velvetg out-dir [-ins_length 260] | tee logfile
Please check that "out-dir/meta-velvetg.contigs.fa" file is created. The FASTA file is the main assembling results.
Advanced topics 1: Manual setting of k-mer coverage peaks (>= 1.1.01)
Contrary to single-genome assemblers (e.g., Velvet and SOAPdenovo), MetaVelvet assumes metagenomics settings. Accordingly, k-mer coverage histogram might be multi-modal rather than uni-modal. Please note that the coverage peak parameters can largely affect MetaVelvet assembling results. Although simple and automatic peak detection algorithm is implemented in meta-velvetg, we strongly recommend manual inspection of k-mer coverage peaks and manual setting of the coverage peak parameters. For manually setting the coverage peak parameters, please execute the following procedures.
- Execute velveth, velvetg, and meta-velvetg as in the "Getting started" section. Please check that "out-dir/meta-velvetg.Graph2-stats.txt" is created.
- Draw a k-mer coverage histogram and manually determine the k-mer coverage peaks.
~$ R
Please determine the coverage peak values from the histogram.
(R) > install.packages("plotrix")
(R) > library(plotrix)
(R) > data = read.table("out-dir/meta-velvetg.Graph2-stats.txt", header=TRUE)
(R) > weighted.hist(data$short1_cov, data$lgth, breaks=seq(0, 200, by=1))
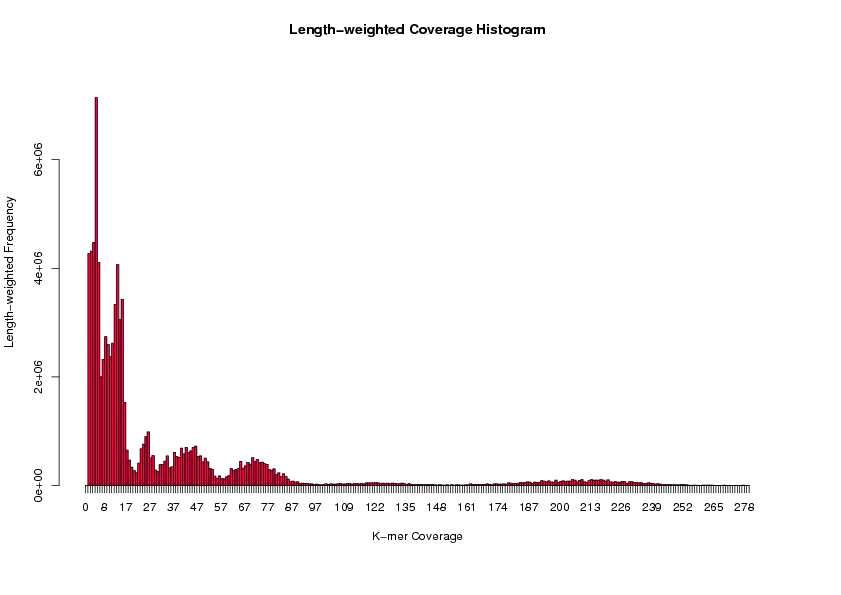
For example, from the above histogram, seven coverage peaks are observed (around 210x, 120x, 70x, 45x, 23x, 12x, and 6x).
If errors are occurred in this step, please see the "Trouble shootings" section. - Run meta-velvetg with manual setting of coverage peaks.
~$ meta-velvetg out-dir -ins_length 260 \
Please note that meta-velvetg assumes that the peak values are sorted in a descending order.
-exp_covs 214_122_70_43_25_13.5
Advanced topics 2: Use Bambus2 scaffolding module (>= 1.1.01)
Bambus2 is a scaffolding module that can be applicable to metagenomics settings. MetaVelvet uses a novel graph splitting algorithm during contiging process, and uses the scaffolding module of Velvet (RockBand and Pebble) during scaffolding process. Alternatively, users can output MetaVelvet contigs and uses Bambus2 scaffolding module instead of using RockBand and Pebble.
To install Bambus2 programs, please see their web site http://www.cbcb.umd.edu/software/bambus/.
- Execute velveth, velvetg as in the "Getting started" section. Please check that "out-dir/Sequences", "out-dir/Roadmaps", and "out-dir/Graph2" files are created.
- Execute meta-velvetg with -amos_file and -scaffolding options.
~$ meta-velvetg out-dir -ins_length 260 \
Please check that "out-dir/meta-velvetg.asm.afg" file is created. The file is meta-velvetg contiging result in an AMOS format.
-exp_covs 214_122_70_43_25_13.5 \
-amos_file yes -scaffolding no - Create amos bank.
~$ bank-transact -m out-dir/meta-velvetg.asm.afg -b bank-dir -cf
- Use the paired-end information to construct a collection of contig links
~$ clk -b bank-dir 2> /dev/null
- Bundle the contig links into a collection of contig edges
~$ Bundler -b bank-dir -t M 2> /dev/null
- Identify genomic repeats and output them
~$ MarkRepeats -b bank-dir \
-redundancy num-pairs \
2> /dev/null \
| tee repeat-file - Order and orient contigs according to repeat and link information
~$ OrientContigs -b bank-dir \
-redundancy num-pairs \
-prefix output-prefix \
-repeats repeat-file \
-all \
2> /dev/null - Linearize the scaffolds
~$ untangle -e output-prefix.evidence.xml \
-s output-prefix.out.xml \
-o output-prefix.untangle.xml \
2> /dev/null - Output contig FastA file
~$ bank2fasta -d -b bank-dir \
| tee contig-file - Output scaffold FastA file
~$ printScaff -merge -e output-prefix.evidence.xml \
Then, scaffold-file is the final scaffolding results in the FastA format.
-o output-prefix \
-s output-prefix.untangle.xml \
-l output-prefix.library \
-f scaffold-file \
2> /dev/null
Advanced topics 3: How to use paired-end information for graph decomposition (>= 1.2.01)
In order to decompose a complecated metagenomic de Bruijn graph into simpler subgraphs,
MetaVelvet searches the following X-structure, i.e., chimeric nodes have two incoming edges and two outgoing edges.
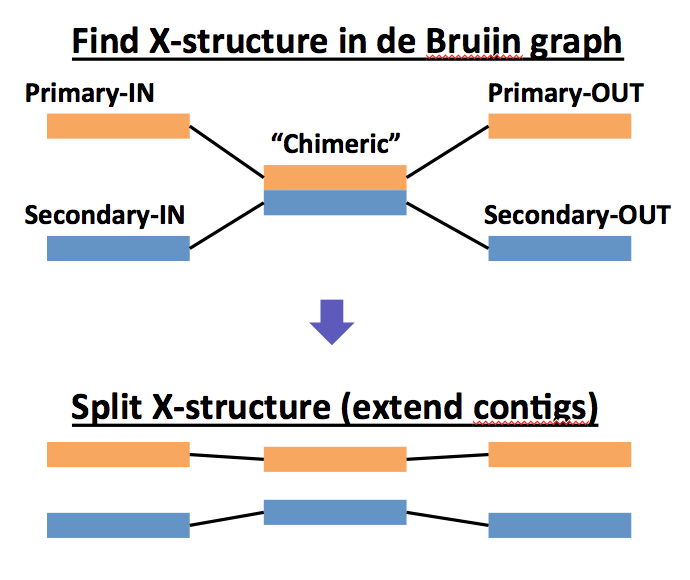
Then, MetaVelvet split the X-structures when the following conditions are satisfied:
- Primary-IN and Primary-OUT nodes are classified into the same peak.
- Secondary-IN and Secondary-OUT nodes are classified into the same peak.
- Chimeric node has a coverage value mostly equal (within 50% difference by default) to the average between the sum of coverage values of the two origin nodes of incoming edges (Primary-IN and Secondary-IN), and the sum of the two destination nodes of outgoing edges (Primary-OUT and Secondary-OUT).
In order to accurately avoid chimeric contigs/scaffolds caused by subgraph decomposition, we added the following two conditions (>= 1.2.01):
- The number of consistent paired-end connections is equal to or larger than a certain value. This cutoff value can be specified by the -valid_connections option (default: 1).
- The number of inconsistent paired-end connections is equal to or smaller than a certain value. This cutoff value can be specified by the -noise_connections option (default: 0).
- For lower compatibility, users can turn off the paired-end conditions by the -use_connections option.
Based upon our accuracy evaluation, -valid_connections 1 -noise_connections 0 is an appropriate setting when very similar species co-exist in an environment (MetaVelvet can efficiently avoid misassemblies). Otherwise, -valid_connections 0 -noise_connections 0 is a more appropriate setting (MetaVelvet can achieve a greater contig/scaffold N50 while avoiding misassemblies).
Frequently asked questions
- Q: meta-velveth is not generated in the new version (>= 1.1.01). Is it problem?
A: This is not problem. The usage of MetaVelvet is changed when the version 1.1.01 is released, and the new version does not include meta-velveth. Instead, please use velveth, velvetg, and meta-velvetg. - Q: When only one coverage peak is detected (or manually input), is there any difference between MetaVelvet and Velvet algorithms?
A: There is no substantial difference. In such cases, meta-velvetg moves to "single-peak mode" and graph splitting functions in meta-velvetg is not called. Instead of graph splitting functions, standard velvet functions are called in such cases. -
Q: What's the difference in working procedures between velvetg, meta-velvetg (<= 0.3.1), and meta-velvetg (>= 1.1.01)?
A: The following is the working procedure of velvetg, meta-velvetg (<= 0.3.1), and meta-velvetg (>= 1.1.01):
velvetg & meta-velvetg(<=0.3.1) :
Load Sequences & Roadmaps file
-> Generate PreGraph file
-> Generate Graph or Graph2 file
-> Generate contigs.fa and LastGraph
meta-velvetg (>= 1.1.01):
Load Sequences & Roadmaps & Graph2 file
-> Generate meta-velvetg.contigs.fa and meta-velvetg.LastGraph
- Q: Is version compatibility between Velvet and MetaVelvet fully tested?
A: Version compatibility between Velvet-1.0.06 and MetaVelvet-1.1.01 is fully tested.
Trouble shootings
- Trouble: When drawing k-mer coverage histogram (as in the "Advanced topics 1" section),
the following warning messages is appeared:
> weighted.hist(data$shot1_cov,data$lgth,breaks=seq(0,200,1))
Solution: This warning (error) is caused by "Inf" values in the Graph2 node stats. Accordingly, by removing "Inf" values from the Graph2 stats, the error is resolved:
Warning messages:
1: In min(x, na.rm = na.rm) :
no non-missing arguments to min; returning Inf
2: In max(x, na.rm = na.rm) :
no non-missing arguments to max; returning -Inf
3: In weighted.hist(data$shot1_cov, data$lgth, breaks = seq(0, 200, :
Areas will not relate to frequencies
$ head -n 1 meta-velvetg.Graph2-stats.txt \
> meta-velvetg.Graph2-stats.rmInf.txt
$ perl -ne '{print $_ unless /Inf/;}' \
meta-velvetg.Graph2-stats.txt \
>> meta-velvetg.Graph2-stats.rmInf.txt
$ R
> library(plotrix)
> data = read.table("meta-velvetg.Graph2-stats.rmInf.txt", header=TRUE)
> weighted.hist(data$shot1_cov,data$lgth,breaks=seq(0,200,1))